
PCSK9 : DU GÈNE À LA FONCTION
La génétique a une place particulière dans l’histoire de PCSK9. L’identification de PCSK9 s’est faite en 2003 par le groupe de Catherine Boileau dans des familles françaises ayant une hypercholestérolémie familiale et ne présentant pas de mutations du gène du recepteur au LDL (LDLR) ou de l’apolipoproteine B (apoB). (1)
PCSK9 est principalement exprimée au niveau du foie, de l’intestin et du rein. La seule activité biologique clairement reconnue de PCSK9 est celle d’inhiber le LDLR. La surexpression adénovirale de PCSK9 in vivo chez la souris entraine une augmentation des concentrations plasmatiques de cholestérol, liée à une diminution de l’expression hépatique du LDLR.(5) Des expériences chez la souris montrent que PCSK9 est exclusivement sécrétée par le foie. En effet, on ne retrouve pas de PCSK9 dans le plasma chez les souris spécifiquement invalidées pour PCSK9 dans le foie.(8)
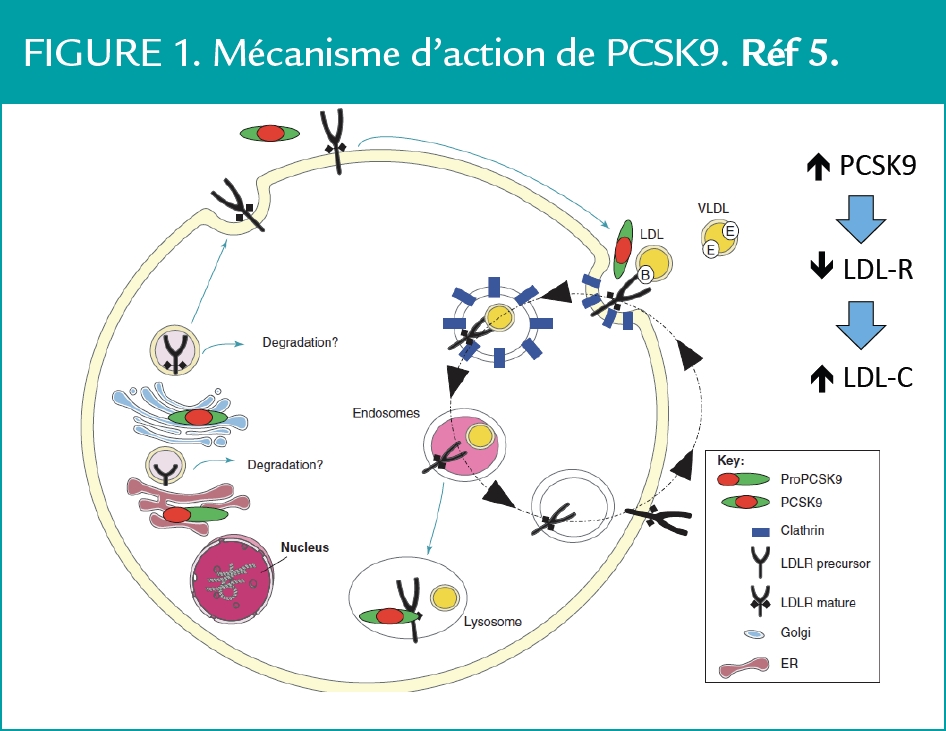
PCSK9 a une demie vie courte (5 min) et agit très rapidement sur le LDLR. PCSK9 agit comme une véritable hormone et module l’expression du LDLR dans d’autres organes dont l’intestin, le tissu adipeux, l’îlot de Langerhans, les poumons, les reins. Pour résumer, en l’absence de PCSK9, la lipoprotéine LDL se lie au LDLR via l’apoB et l’ensemble est endocytosé dans l’hépatocyte. Alors que la particule de LDL est orientée vers les lysosomes où ses composants sont métabolisés ou recyclés, le LDLR est redirigé vers la surface de l’hépatocyte pour lier et épurer une nouvelle particule de LDL. Le LDLR peut être ainsi recyclé 150 fois ; PCSK9 circulante interrompt ce cycle en se liant au domaine extracellulaire EGF-A du LDLR, agissant ainsi comme une protéine chaperone. En effet, il est important de préciser ici que l’activité catalytique de PCSK9 n’est pas nécessaire à son action inhibitrice vis à vis du LDLR.
L’ensemble PCSK9-LDLR est endocytosé, mais au lieu de repartir vers la membrane, le LDLR et PCSK9 sont détruits dans les lysosomes (Figure 1). En résumé, plus les concentrations plasmatiques de PCSK9 sont élevées, moins la quantité de LDLR présent à la surface de l’hépatocyte est importante et plus la cholestérolémie augmente.
A l’heure actuelle, il existe deux grandes classes d’inhibiteurs de PCSK9 en cours d’évaluation clinique, tous en préparations injectables :
Les anticorps monoclonaux ciblent la protéine circulante afin d’empêcher son interaction avec le LDLR et deux disposent à l’heure actuelle d’une AMM européenne et américaine : alirocumab (Praluent®, Sanofi-Regeneron) et evolocumab (Repatha®, Amgen). Ces anticorps monoclonaux sont humanisés à 100% et la fréquence des injections sous-cutanées varie de 2 à 4 semaines : dose de 75 mg ou 150 mg tous les 15 jours pour l’alirocumab et dose de 140 mg tous les 15 jours ou 420 mg tous les mois pour l’evolocumab. De façon rassurante, les résultats obtenus sont comparables avec les 2 molécules (baisse du LDL-cholestérol d’environ 50-60%), suggérant un effet classe des anticorps monoclonaux anti-PCSK9.(9)
Les SiRNA entraînent une dégradation de l’ARNm de PCSK9 ou de sa traduction et donc la diminution de sa synthèse protéique. Un essai de phase 1 a eu lieu chez l’homme avec les SiRNA anti-PCSK9 (Alnylam) avec une diminution de 68% des concentrations de PCSK9 et de 41% du LDL-C à la dose maximale. Il est prématuré de prédire l’efficacité relative de ces 2 stratégies (anticorps monoclonaux vs SiRNA), même si les premières données de tolérance sont en faveur des anticorps monoclonaux. Un des avantages théoriques des SiRNA est qu’ils pourraient également inhiber l’action intracellulaire de PCSK9, qui demeure mal connue.
Moins de 10 années se sont écoulées entre la découverte de PCSK9 en 2003 et les premiers essais thérapeutiques avec les anticorps monoclonaux anti-PCSK9 en 2012, faisant de PCSK9 une véritable « success story » de la recherche translationnelle appliquée.
Bertrand Cariou, Nantes
Liens d’intérêts : B. Cariou déclare des interventions ponctuelles (essais cliniques, conseils, conférences, colloques et actions de formation) avec les laboratoires AMGEN et SANOFI-REGENERON et avoir été pris en charge à l’occasion de congrès par AMGEN et SANOFI-REGENERON.
RÉFÉRENCES
1. Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003 ; 34 : 154-6
2. Marduel M, Carrié A, Sassolas A, et al. Molecular spectrum of autosomal dominant hypercholesterolemia in France. Hum. Mutat. 2010 ; 31 : E1811-24
3. Cohen JC, Boerwinkle E, Mosley TH, et al. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006 ; 354 : 1264-72
4. Ference BA, Yoo W, Alesh I, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease : a Mendelian randomization analysis. J Am Coll Cardiol 2012 ; 60 : 2631-9
5. Maxwell KN, Breslow JL. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc. Natl. Acad. Sci. USA. 2004 ; 101 : 7100-5
6. Rashid S, Curtis DE, Garuti R, et al. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc. Natl. Acad. Sci. USA. 2005 ; 102 : 5374-9
7. Lagace TA, Curtis DE, Garuti R, et al. Secreted PCSK9 decreases the number of LDL receptor in hepatocytes and in livers of parabiotic mice. J. Clin. Invest. 2006 ; 116 : 2995-3005
8. Zaid A, Roubtsova A, Essalmani R, et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9): hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology 2008; 48:646-54.
9. Farnier M. PCSK9 inhibitors. Curr Opin Lipidol 2013; 24: 251-8.
_______________________________________________________
HYPERCHOLESTÉROLÉMIE FAMILIALE, MODÈLE DE PATIENT À HAUT RISQUE CARDIO-VASCULAIRE
L’hypercholestérolémie familiale (HF) est une maladie génétique peu diagnostiquée et insuffisamment traitée en France, du moins dans sa forme hétérozygote (He).
C’est pourtant une maladie importante à connaitre car l’exposition au long cours à un taux élevé de LDL-cholestérol (LDL-C) induit un risque accru de maladie cardiovasculaire liée à l’athérosclérose. La forme homozygote, exceptionnelle est prise en charge en milieu spécialisé compte-tenu de sa sévérité.
Le diagnostic de HeHF peut être évoqué dans l’une des circonstances suivantes : (1,2)
– concentration élevée de LDL-C c’est-à-dire, en l’absence de traitement, supérieur à 1,9 g/l (1,9) chez l’adulte et supérieur à 1,6 g/l chez l’enfant/adolescent; (1,6)
– présence de dépôts extravasculaires de cholestérol, en particulier de xanthomes tendineux;
– parents déjà connus comme porteurs de HF;
– notion d’accidents vasculaires précoces personnels et familiaux.
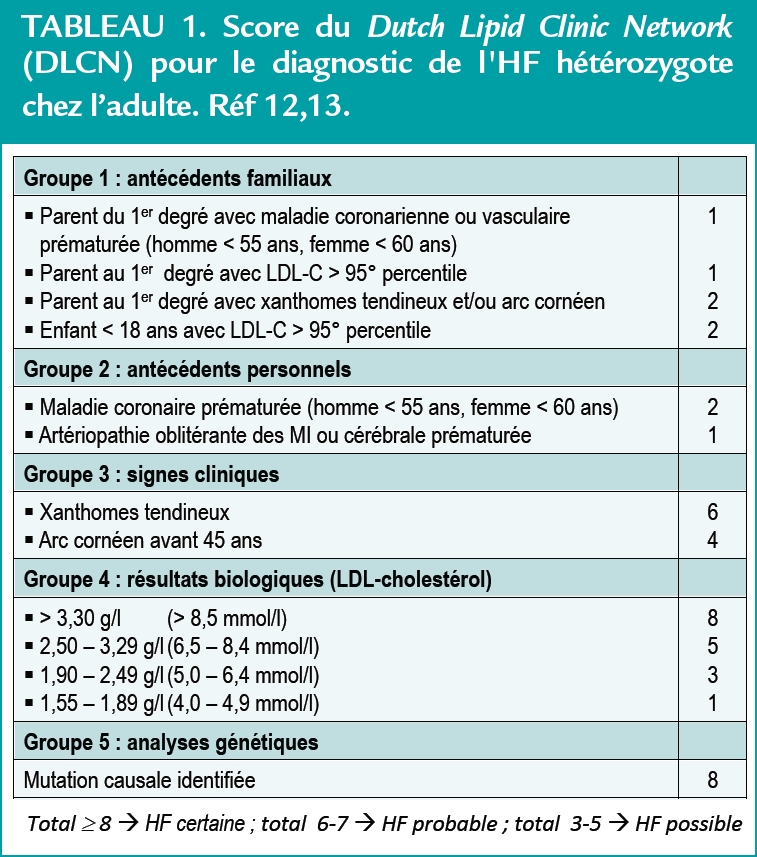
Deux approches sont possibles pour identifier une HF hétérozygote, l’utilisation de grilles de score construites à partir de critères cliniques et biologiques ou un diagnostic génétique.(2,3) La grille de score la plus répandue est issue du “Dutch Lipid Clinic Network (DLCN)” qui utilise 4 groupes de critères cliniques et/ou biologiques pour obtenir un diagnostic “certain” ou “probable”.
Un score “possible” est beaucoup moins pertinent pour identifier une HF. L’analyse génétique positive donne bien sûr un diagnostic de certitude (Tableau 1).
Lorsqu’un patient avec HF est identifié, il est indispensable de déclencher une enquête familiale avec dépistage en cascade. Ce dépistage en cascade est plus efficient qu’un dépistage systématique dans la population générale est doit être réalisé chez tous les parents du 1er degré de HF diagnostiquée. Il nécessite au minimum un bilan lipidique et, lorsque cela est possible, la réalisation de l’analyse génétique dans les cas où la mutation a été identifiée pour le cas index.
L’HeHF, maladie autosomique dominante, n’est pas une maladie génétique rare : sa prévalence a longtemps été estimée à 1/500 naissances, mais les données récentes sont en faveur d’une prévalence aux alentours 1/250 naissances.(1,4) De plus, compte-tenu du risque cardiovasculaire accru, la prévalence est plus élevée dans des cohortes de patients coronariens : dans l’étude EUROASPIRE IV,(5) la prévalence des patients avec HF potentielle a été trouvée de 8,3% et atteignait 20% environ chez ceux avec maladie coronaire précoce (survenue avant 50 ans). De même, dans une population de patients hospitalisés pour syndrome coronaire aigu en Suisse, la prévalence de patients avec HF probable ou certaine est proche de 5% chez les patients avec syndrome coronarien aigu prématuré (inférieur à 55 ans chez l’homme, ou 60 ans chez la femme). (6)
Sans traitement, un patient avec HF a un risque de maladie coronaire environ 13 fois plus élevé que dans la population générale.(1) Le traitement par statine ou par statine/ézétimibe a réduit le risque cardiovasculaire de ces patients et le risque résiduel sous traitement dépend de façon étroite de l’âge du diagnostic et de début du traitement, des facteurs de risque cardiovasculaire associés et de la sévérité de l’HeHF. Dans une large cohorte récente de patients avec HF génétiquement diagnostiquée suivis en Norvège de 1992 à 2010, le risque de mortalité cardiovasculaire restait malgré le traitement 2 fois plus élevé chez les hommes et 3 fois plus élevé chez les femmes par comparaison à la population générale.(7)
Le traitement médicamenteux d’une HeHF repose en priorité sur les statines de forte intensité et à fortes doses (essentiellement atorvastatine et rosuvastatine). La plupart des patients n’étant pas à l’objectif sous statine en monothérapie, une association avec ézétimibe est fréquemment nécessaire. Les objectifs thérapeutiques proposés dans les recommandations de la Nouvelle Société Française d’Athérosclérose (NSFA) sont indiquées dans le tableau 2.(2) Avec un traitement bien conduit, il est possible d’abaisser le LDL-C de l’ordre de 50 à 70%, sans toutefois pouvoir atteindre les objectifs recommandés vue la sévérité d’un grand nombre de formes familiales.

De ce fait, d’autres options thérapeutiques doivent être envisagées et parmi celles-ci, ce sont les inhibiteurs de PCSK9 et les inhibiteurs de la CETP qui ont fait l’objet d’études spécifiques aux formes familiales hétérozygotes. Ainsi les deux inhibiteurs de PCSK9, alirocumab et evolocumab, récemment approuvés en Europe, sont efficaces pour abaisser le LDL-C avec, par rapport au placebo, des diminutions du LDL-C de l’ordre de 50 à 60%.(8,9) Dans ces études, les inhibiteurs de PCSK9 se sont révélés bien tolérés, mais l’effet sur la morbi-mortalité cardiovasculaire reste à démontrer.
Michel Farnier, Dijon
Liens d’intérêts : Le docteur Michel Farnier déclare avoir reçu des honoraires en tant qu’investigateur, expert scientifique et/ou conférencier de la part des firmes suivantes : Abbott/Mylan, Amgen, AstraZeneca, Eli Lilly, Genzyme, Kowa, Merck and Co, Pfizer, Roche, Sanofi/Regeneron et Servier.
RÉFÉRENCES
1. Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease. Eur Heart J 2013; 34: 3478-90.
2. Farnier M, Bruckert E, Boileau C, Krempf M. Diagnostic et traitement des hypercholestérolémies familiales (HF) chez l’adulte: recommandations de la Nouvelle Société Française d’Athérosclérose (NSFA). Presse Med 2013; 42: 930-50.
3. Hovingh GK, Davidson MH, Kastelein JJP, O’Connor AM. Diagnosis and treatment familial hypercholesterolaemia. Eur Heart J 2013; 34: 962-71.
4. Sjouke B, Kusters DM, Kindt I, et al. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalence, genotype-phenotype relationship, and clinical outcome. Eur Heart J 2014; 36: 560-5.
5. De Backer G, Besseling J, Chapman J, et al. on behalf of the EUROASPIRE Investigators. Prevalence and management of familial hypercholesterolaemia in coronary patients: an analysis of EUROASPIRE IV, a study of the European Society of Cardiology. Atherosclerosis 2015; 241: 169-75.
6. Nanchen D, Gencer B, Auer R, et al. Prevalence and management of familial hypercholesterolaemia in patients with acute coronary syndromes. Eur Heart J 2015; 36: 2438-45.
7. Mundal L, Sarancic M, Ose L, et al. Mortality among patients with familial hypercholesterolemia: a registry-based study in Norway, 1992-2010. J Am Heart Assoc 2014; 3:e001236 doi/10.1161
8. Kastelein JJP, Ginsberg HN, Langslet G, et al. ODYSSEY FH I and FH II: 78-week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolemia. Eur Heart J 2015; 36: 2996-3003
9. Raal FJ, Stein EA, Dufour R, et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolemia (RUTHERFORD-2): a randomized, double-blind, placebo-controlled trial. Lancet 2015; 385: 331-40.
_______________________________________________________
INHIBITEURS PCSK9, ENSEIGNEMENTS CLÉS DES PROGRAMMES DE DÉVELOPPEMENT CLINIQUE
Les mutations de PCSK9 avec gain de fonction sont caractérisées par un LDL-C élevé et une forte incidence de maladies CV. L’effet à long terme (15 ans) d’une perte de fonction a été bien étudié avec l’étude ARIC.(1)
• Chez 3 363 sujets de race noire, où sa haute prévalence est connue, une mutation PCSK9 de type “non-sens” (perte de fonction) était présente chez 2,6% avec des réductions moyennes de 28% du LDL-C et de 88% (p = 0,03) du risque des événements CV (IDM, coronaropathie fatale ou revascularisation coronaire).
• Chez 9 524 sujets de race blanche, 2 mutations de type “variation” connues comme très prévalentes, ont été trouvées dans 3,2% des cas avec des réductions moyennes de 47% du LDL-C et de 50% (p = 0,003) du risque d’événements CV. Ainsi, une diminution, même modérée, du LDL-C, provoquée par la suppression d’activité PCSK9 peut, sur une vie, donner lieu à des réductions majeures du risque CV.
Un tel blocage est particulièrement intéressant en association avec les statines dont l’un des effets est de stimuler PCSK9. Quatre anticorps monoclonaux humains, injectables en SC, sont à des stades divers de développement. Les phases 3 de l’évolocumab et l’alirocumab sont achevées. Ainsi, il aura suffi d’une quinzaine d’années depuis la découverte de CPSK9 pour compléter des programmes cliniques qui totalisent à ce jour plus de 70 000 patients.
Une méta-analyse de 2014 (N = 38 153) montre qu’en monothérapie, même avec de fortes doses de statines (pravastatine 40mg/j ou rosuvastatine 20 mg/j), 40% des patients n’atteignent pas la cible thérapeutique (50% de la valeur initiale).(2)
Une précédente (2013, 1,9 millions de patients), soulignait que l’impact de la non-adhérence au traitement est plus important avec les statines qu’avec les autres traitements de prévention.(3) Avec les anti-PCSK9, en monothérapie, les études de phase II, en particulier de l’alirocumab (150 mg toutes les 2 semaines) ont bien montré que l’effet est dose-dépendant et qu’il est possible, d’obtenir une réduction parfaitement stable du LDL-C (de 50 à 60%).(4)
En association à 10 ou 80 mg d’atorvastatine, le niveau de LDL-C atteint sous alirocumab est identique et maintenu de façon stable.(5)
Dans les études de phase III, avec alirocumab ou évolocumab, dans lesquelles des patients non à leur objectif recevaient soit un traitement standard à dose maximum tolérée, soit le même traitement avec un anti-PCSK9 les investigateurs ont eu, outre la confirmation d’une baisse supplémentaire du LDL-C, un fort signal en faveur d’une diminution de la morbimortalité CV. Il faudra attendre la confirmation de ce bénéfice dans les études de phase III spécifiquement dédiées.
En pratique, les anti-PCSK9 devraient tirer leurs meilleures indications chez les patients avec HeHF (Figure 1) ou lorsque les statines sont insuffisantes (Goal-Inhibiting Statin Resistance ou GISR)(2) ou mal tolérées (Goal-Inhibiting Statin Intolerance ou GISI).(6,7) L’alirocumab comme l’évolocumab ont démontré leur efficacité pour les HeHF, notamment dans les études ODYSSEY FH1/FH2(8) et RUTHERFORD.(9)
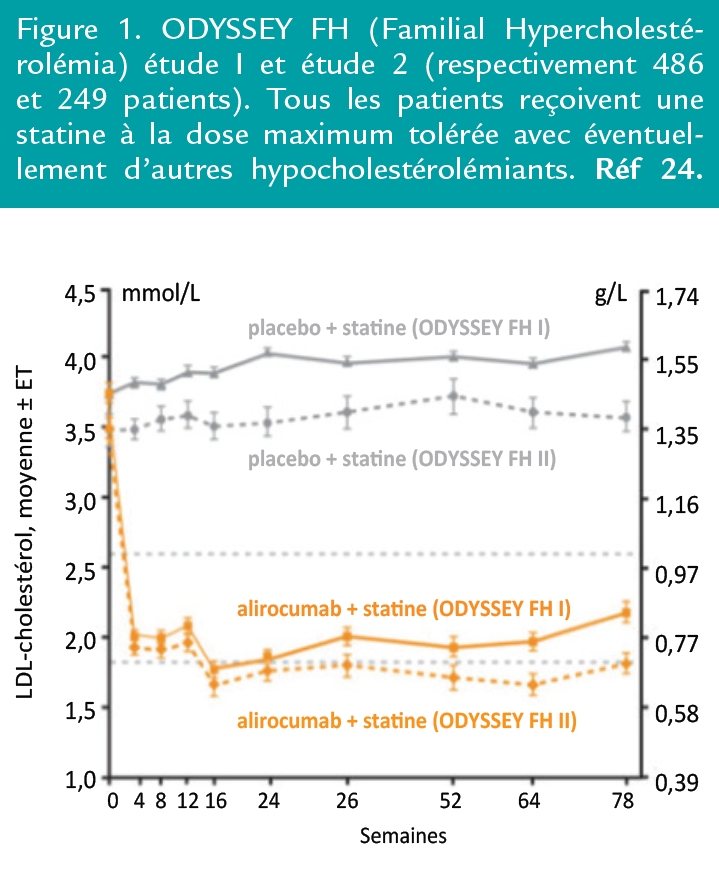
Une possible intolérance musculaire aux statines doit être évaluée de façon rigoureuse(10) et un algorithme décisionnel a été proposé par la Société Européenne d’Athérosclérose.(11) Les données récentes de l’étude ODYSSEY-alternative ont montré qu’en substitution d’un traitement par statine mal toléré, l’alirocumab permet une baisse plus importante du LDL-C que l’ézétimibe et avec une meilleure tolérance.(12)
La question des risques liés à des abaissements importants du LDL-C mérite d’être posée : les données de certaines études réalisées avec des statines puissantes à forte dose (comme JUPITER) sont pour l’instant rassurantes.(13) Celle d’éventuels désordres neurocognitifs suscite la plus grande attention mais il faudra attendre l’accumulation des données de tolérance des autres études de phase III avec les anti-PCSK9 pour avoir une opinion définitive.
En conclusion, l’efficacité (et sans doute la tolérance) remarquable des anti-PCSK9 a remis l’accent sur des problèmes essentiels qui limitaient la prise en charge du risque lipidique.
François Schiele, Besançon
RÉFÉRENCES
1. Cohen JC, Boerwinkle E, Mosley TH, Jr., Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006;354:1264-72.
2. Boekholdt SM, Hovingh GK, Mora S, et al. Very low levels of atherogenic lipoproteins and the risk for cardiovascular events: a meta-analysis of statin trials. J Am Coll Cardiol 2014;64:485-94.
3. Chowdhury R, Khan H, Heydon E, et al. Adherence to cardiovascular therapy: a meta-analysis of prevalence and clinical consequences. Eur Heart J 2013;34:2940-8.
4. Stein EA, Gipe D, Bergeron J, et al. Effect of a monoclonal antibody to PCSK9, REGN727/ SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet 2012;380:29-36.
5. Roth EM, McKenney JM, Hanotin C, Asset G, Stein EA. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N Engl J Med 2012;367:1891-900.
6. Bruckert E, Hayem G, Dejager S, Yau C, Begaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients–the PRIMO study. Cardiovasc Drugs Ther 2005;19:403-14.
7. Mancini GB, Baker S, Bergeron J, et al. Diagnosis, prevention, and management of statin adverse effects and intolerance: proceedings of a Canadian Working Group Consensus Conference. Can J Cardiol 2011;27:635-62.
8. Kastelein JJ, Ginsberg HN, Langslet G, et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J 2015;36:2996-3003.
9. Raal FJ, Stein EA, Dufour R, et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebocontrolled trial. Lancet 2015;385:331-40.
10. Rosenson RS, Baker SK, Jacobson TA, Kopecky SL, Parker BA, The National Lipid Association’s Muscle Safety Expert P. An assessment by the Statin Muscle Safety Task Force: 2014 update. J Clin Lipidol 2014;8:S58-71.
11. Stroes ES, Thompson PD, Corsini A, et al. Statin-associated muscle symptoms: impact on statin therapy-European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur Heart J 2015;36:1012-22.
12. Moriarty PM, Thompson PD, Cannon CP, et al. Efficacy and safety of alirocumab vs ezetimibe in statin-intolerant patients, with a statin rechallenge arm: The ODYSSEY ALTERNATIVE randomized trial. J Clin Lipidol 2015;9:758-69.
13. Hsia J, MacFadyen JG, Monyak J, Ridker PM. Cardiovascular event reduction and adverse events among subjects attaining low-density lipoprotein cholesterol <50 mg/dl with rosuvastatin. The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). J Am Coll Cardiol 2011;57:1666-75.
Article publié dans le supplément du Cordiam N°12 (Juin 2016)