
Au sein d’une patientèle on peut estimer que sur 20 patients qui ont une hypercholestérolémie, un seul présente une forme familiale définie par le caractère autosomique dominant. Chez le cardiologue cette fréquence peut être plus élevée en raison du biais lié au risque coronaire. C’est autour de ce concept que se construit le diagnostic. C’est à cause de ces caractéristiques et en particulier du très haut risque vasculaire que se justifie une prise en charge spécifique.
Autosomique dominant c’est la certitude d’un risque cardiovasculaire considérable.
Cette forme est en effet caractérisée par la sévérité de l’augmentation du LDL-cholestérol, par la fréquente augmentation de la Lp(a), par le HDL dysfonctionnel et surtout par une exposition depuis la naissance à un cholestérol élevé. L’étude de Copenhague faite dans une grande population démontre que les patients atteints de forme familiale ont un risque de maladie coronaire multiplié par 13. D’autres études confirment ce risque très élevé (revue in 1). De façon intéressante une vaste
étude génétique montre que chez les sujets ayant eu un infarctus du myocarde il y a 13 fois plus de mutation dite disruptive du gène du récepteur aux LDL(2).
Compte tenu du fait que « statistiquement » une personne sur deux est atteinte dans la famille, il existe souvent une histoire de maladie cardiovasculaire précoce.
La survenue d’un syndrome coronaire aigu peut être précoce (20 ans) surtout chez le fumeur. C’est donc une faute thérapeutique de ne pas traiter ces patients jeunes.
Autosomique dominant c’est un LDL-cholestérol très élevé depuis la naissance avec une possibilité de dépôt extravasculaires
Plus il est élevé plus la probabilité d’avoir une forme familiale est élevée. Un signe d’alerte facile pour le médecin c’est un LDL-cholestérol supérieur à 1.90 g/l.
Dans la forme familiale le niveau de cholestérol dans le sang est en moyenne le double de la normale.
Dans les autres formes appelées polygéniques le niveau est le plus souvent plus bas. Un Français sur 500 a une hypercholestérolémie familiale alors qu’on peut considérer que au moins un tiers des français a une hypercholestérolémie polygénique (mélange de prédisposition génétique, de diététique non adaptée ou parfois de surpoids). L’augmentation du LDL-c est en général stable tout au long de la vie.
L’élévation du LDL-c existe depuis la naissance. En pratique cela veut dire que si on retrouve une valeur de LDL-c normale ou subnormale quand le patient était jeune, cela plaide fortement contre le diagnostic.
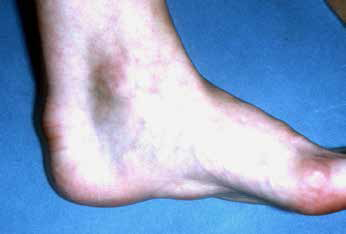
Figure 1 : Xanthomes tendineux
Les dépôts extravasculaires de cholestérol sont fréquents et comprennent l’arc cornéen (évoque une hypercholestérolémie familiale quand il est présent avant 45 ans), le xanthélasma, les xanthomes tendineux (figure 1). Ils sont le plus souvent visibles et palpables sur les tendons d’Achille et sur les tendons extenseurs des doigts de la main. Les xanthomes tendineux sont pratiquement pathognomoniques de forme familiale. Ils sont un marqueur de l’exposition prolongée à un LDL-c très élevé. Les raisons qui font que même dans les formes polygéniques sévères on ne retrouve pas de xanthomes, sont mal connues.
Autosomique dominant c’est une transmission familiale bien caractéristique.
L’histoire familiale doit être compatible avec une forme autosomique dominante c’est-à-dire qu’en pratique la moitié de la famille est porteur d’hypercholestérolémie avec le plus souvent des augmentations similaires à celle du parent atteint.
Il faut insister sur le fait que dans les formes polygéniques il peut aussi y avoir des antécédents familiaux d’hypercholestérolémie mais parfois ce sont les deux parents, souvent les niveaux de LDL-c sont variables au sein de la famille, les enfants sont habituellement normocholestérolémiques.
Les scores cliniques
Les scores cliniques sont un résumé de ces caractéristiques.
Le diagnostic qui peut se faire sur un score clinique et biologique (échelle développée en Hollande, Tableau 1) peut aussi être fait ou confirmé par l’analyse génétique (cf infra). Ce score met en « musique » sous forme mathématique les caractéristiques de la forme familiale.

Le diagnostic différentiel
Il faut bien entendu vérifier qu’il n’existe pas une cause autre d’hypercholestérolémie (exemple une hypothyroïdie).
Les formes polygéniques
L’augmentation du LDL-cholestérol dans le sang est liée à des facteurs génétiques et des facteurs de l’environnement comme la diététique riche en cholestérol ou acides gras saturés, la prise de poids et le vieillissement. La majorité des hypercholestérolémies primaires est appelée polygénique car elles sont la conséquence d’une somme variable d’anomalies génétiques qui quand elles sont isolées ont un impact modeste. Ces formes polygéniques ont donc une sévérité variable en fonction du nombre d’anomalies présentes. Elles ont toutefois comme caractéristique principale l’apparition à l’âge adulte et l’aggravation avec le temps. Les antécédents familiaux de dyslipidémie sont donc aussi très variables (aucun, un ou deux parents avec hypercholestérolémie).
Le diagnostic génétique
Les anomalies génétiques responsables d’hypercholestérolémie familiales touchent le plus souvent le récepteur des LDL. En France les mutations sur ce récepteur expliquent 80% des formes familiales. Il existe plus de 1000 mutations décrites. Certaines entrainent une absence totale du récepteur ou de sa fonction et s’accompagnent d’une forte élévation du cholestérol. D’autres laissent persister une petite partie de la fonction de la protéine et sont un peu moins sévères. La mutation qui touche l’apolipoprotéine B est plus rare et touche quelque pourcent des patients.
Les mutations qui touchent la protéine PCSK9 sont très rares avec seulement quelques familles touchées. Il reste des formes familiales typiques pour lesquelles la mutation n’est pas identifiée. Le diagnostic génétique est possible dans les laboratoires agréés. Il en existe 4 en France.
En pratique pour faire le diagnostic génétique il faut soit adresser le patient dans un centre spécialisé soit faire parvenir un tube de sang avec le consentement adapté dans un des laboratoires agréés.
Prise en charge de l’hypercholestérolémie familiale
La première raison justifiant la nécessité d’identifier clairement ces patients est que le risque de faire un accident cardiovasculaire jeune est très élevé et que le traitement correctement conduit protège contre ce risque. La deuxième raison est qu’il faut identifier les autres membres de la famille quand un individu est diagnostiqué avec cette forme familiale. Les enfants doivent êtres dépistés à partir de l’âge de 3 ans et traités à partir de l’âge de 10 ans. Enfin la dernière raison est que l’identification et le diagnostic génétique, quand il est fait, conduisent le plus souvent les patients à une meilleure observance des traitements.
La prise en charge d’une hypercholestérolémie familiale comporte systématiquement deux catégories de mesures :
• des mesures dites hygiéno-diététiques associées au traitement des autres facteurs de risque quand ils existent
• des traitements médicamenteux.
Chez l’adulte,
Mesures diététiques recommandées :
• une réduction des apports globaux en matières grasses,
• une réduction de l’apport en graisses saturées,
• et de façon conjointe une réduction des aliments riches en cholestérol.
Les recommandations sont donc les mêmes que pour les hypercholestérolémies polygéniques.
Traitements médicamenteux
Dans l’hypercholestérolémie familiale, la classe la plus importante de médicaments pour réduire le LDLcholestérol est la famille des statines. En pratique compte tenu de l’élévation importante du LDL-c il est le plus souvent nécessaire de recourir aux statines les plus puissantes en particulier l’atorvastatine ou la rosuvastatine. Dans cette forme les recommandations de débuter chez l’adulte par des statines peu puissantes sont déraisonnables (sauf dans les formes les moins sévères).
Les formes classiques vont habituellement nécessiter une statine puissante à forte dose et même une bithérapie.
Avant l’utilisation des statines, il était classique d’utiliser des résines qui sont des substances qui se lient dans l’intestin à des acides biliaires. Ce traitement (colestyramine) est associé à de nombreux effets secondaires digestifs ce qui limite l’acceptabilité pour les patients. La réduction du LDL-cholestérol obtenue avec les résines, est de 15 à 25% en fonction de la dose utilisée.
L’ézétimibe est une autre molécule qui agit au niveau intestinal, mais par un mécanisme différent. L’ézétimibe bloque de façon spécifique l’absorption intestinale du cholestérol qu’il soit d’origine alimentaire ou biliaire. La réduction moyenne du LDL-cholestérol observée sous ézétimibe est de l’ordre 20%. L’ézétimibe doit essentiellement être utilisé en association avec une statine lorsque le traitement par statine ne permet d’abaisser suffisamment le taux de LDL-c.
Chez l’enfant et l’adolescent
Il est actuellement recommandé de proposer un traitement médicamenteux à partir de l’âge de 8 à 10 ans lorsque le taux de LDL-cholestérol reste supérieur à 1.90 g/l, après une période d’au moins 6 mois de mesures hygièno-diététiques. Il est maintenant recommandé de choisir comme traitement de première intention chez l’enfant et l’adolescent une statine, comme chez l’adulte, mais en utilisant la dose efficace la plus faible(4). Un traitement plus précoce peut parfois être justifié dans des formes sévères, en général après avis spécialisé.
Dans la forme particulièrement grave de l’hypercholestérolémie familiale, forme dite homozygote (quand les deux parents ont transmis la maladie), il est possible de proposer la LDL-aphérèse. Ces formes doivent être prises en charge en milieu spécialisé.
Dans l’avenir une nouvelle classe thérapeutique permettra de mieux corriger les formes les plus graves de cette dyslipidémie. Il s’agit des anticorps antiPCSK9.
« L’hypercholestérolémie familiale doit être diagnostiquée car sa prise en charge est spécifique »
Informations pratiques
Pour en savoir plus 3 publications de la société Européenne d’Athérosclérose font le point sur la forme hétérozygote de l’enfant et de l’adulte ainsi que sur la forme homozygote particulièrement grave mais très rare puisqu’elle touche un sujet sur un million(1,3,4).
Enfin il existe une association de malades (voir image de tête – www.anhet.fr). Un registre français a été mis en place pour améliorer les connaissances sur l’histoire naturelle de la maladie et montrer l’évolution avec les nouvelles thérapeutiques(5).
Eric Bruckert
Source : Cordiam n°11
RÉFÉRENCES
(1) Nordestgaard BG, Chapman MJ, Humphries SE, et al European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013 Dec;34(45):3478-90a.
(2) Do R, Stitziel NO, Won HH, Jørgensen AB, et al Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015 Feb 5;518(7537):102-6.
(3) Wiegman A, Gidding SS, Watts GF, et al European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J. 2015 May 25.
(4) Cuchel M, Bruckert E, Ginsberg HN, et al ; European Atherosclerosis Society Consensus Panel on Familial Hypercholesterolaemia. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014 Aug 21;35(32):2146-57.
(5) Béliard S, Carreau V, Carrié A, Giral P, Duchêne E, Farnier M, Ferrières J, Fredenrich A, Krempf M, Luc G, Moulin P, Bruckert E. Improvement in LDL-cholesterol levels of patients with familial hypercholesterolemia: can we do better? Analysis of results obtained during the past two decades in 1669 French subjects. Atherosclerosis. 2014 May;234(1):136-41