
Une AMM européenne a été attribuée au tafamidis (Vyndaqel®) le 17 février dernier pour le traitement de l’amylose cardiaque à transthyrétine (TTR). Ce traitement avait déjà une AMM pour le traitement de la polyneuropathie symptomatique à TTR et est accessible pour les cardiomyopathies TTR en RTU (Recommandation temporaire d’utilisation) depuis novembre 2018. L’objectif de cet article n’est pas d’effectuer une revue exhaustive de l’atteinte cardiaque de l’amylose TTR mais de présenter le contexte général dans lequel le tafamidis a obtenu cette AMM et de discuter de son apport pour la prise en charge de nos patients.
L’amylose TTR : un diagnostic important à faire
Les amyloses à TTR (mutée ou sauvage) et AL peuvent être associées à une atteinte cardiaque. L’amylose héréditaire (ou hATTR), par mutation dans le gène de la transthyrétine, est à transmission autosomique dominante, et touche environ 500 patients en France (1/100 000, âge 30-80 ans, mais 3,4% des afro-américains). L’amylose sénile (wtATTR), à nette prédominance masculine, est assez fréquente (au moins 3/10 000), 1-3% des plus de 75 ans). L’amylose AL (1/100 000, atteinte cardiaque 50%, âge moyen au diagnostic 67 ans), urgence diagnostique et thérapeutique, est liée à la sécrétion de chaînes légères libres d’immunoglobulines monoclonales, produites le plus souvent dans un contexte d’hémopathie maligne (myélome).
Comme beaucoup de pathologies rares, leur diagnostic est difficile parmi les nombreux patients que nous sommes amenés à prendre en charge, mais il est devenu crucial dans la mesure où un traitement spécifique améliorant le pronostic est maintenant disponible. Outre les formes typiques de cardiomyopathies restrictives, les portes d’entrées cardiologiques peuvent être l’insuffisance cardiaque à fraction d’éjection préservée (jusqu’à 13% de wtATTR), le rétrécissement aortique (jusqu’à 16% de wtATTR parmi les patients chirurgicaux) et la cardiomyopathie hypertrophique d’allure sarcomèrique (jusqu’à 5% de hATTR)
Un certain nombre de signes cardiaques peuvent orienter plus spécifiquement vers le diagnostic, tels que microvoltage (20% des ATTR), troubles conductifs de haut degré, hypertrophie ventriculaire gauche concentrique avec éventuellement hypokinésie globale, hypertrophie ventriculaire droite, épanchement péricardique, contraste entre un strain ventriculaire gauche altéré à la base et préservé à l’apex, rehaussement tardif diffus du myocarde en IRM (technique d’inversion-récupération) ou sous-endocardique. Les principales anomalies extracardiaques pouvant renforcer la suspicion sont un syndrome du canal carpien (10% de wtATTR en cas de canaux carpiens opérés après 50 ans chez l’homme) et, surtout pour les formes génétiques, une neuropathie périphérique.
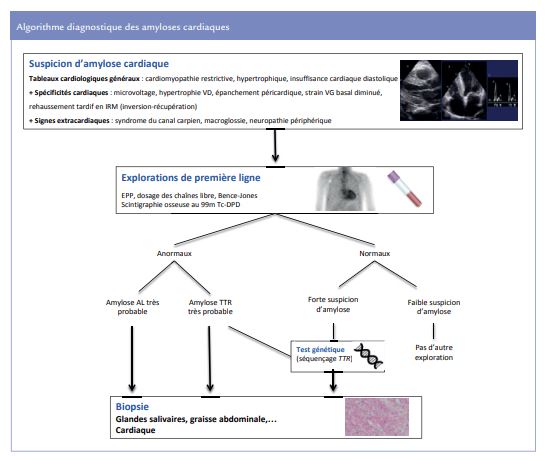
La confirmation du diagnostic d’amylose TTR passe en premier lieu par la scintigraphie osseuse au 99mTc-DPD qui suffi t au diagnostic (spécificité # %) lorsqu’elle montre une franche hyperfixation myocardique (niveaux 3/4 sur une échelle de 0 à 4). Il faut dans tous les cas éliminer une amylose AL, dont le diagnostic repose sur les dosages des chaines légères libres circulantes et l’immunoélectrophorèse des protéines sériques et urinaires (protéinurie de Bence Jones). Le test génétique avec séquençage du gène TTR permet d’affirmer le diagnostic d’amylose héréditaire. Les biopsies extra-cardiaques (glandes salivaires, tissus adipeux, etc.) en première intention, si besoin par des biopsies myocardiques, restent la référence pour le diagnostic positif et le typage. La démarche diagnostique reposant sur ces outils est résumée dans la figure 1.
En l’absence de traitement spécifique, la survie médiane de l’amylose TTR n’est que de 3 à 5 ans après le diagnostic (médianes 3,5 ans si wtATTR et 2,5 ans si hTTR). La prise en charge cardiologique conventionnelle de l’insuffisance cardiaque influe peu sur la survie et doit être adaptée à la pathologie (bêtabloqueurs et inhibiteurs du SRAA à éviter, indications larges des anticoagulants et du pacemaker).
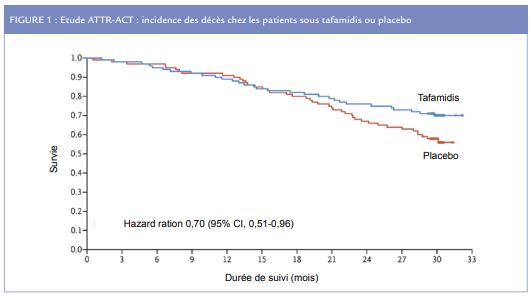
Tafamidis : mécanisme d’action et effet clinique dans l’amylose cardiaque à transthyrétine (étude ATTR-ACT)
La transthyrétine est une protéine de structure tétramérique synthétisée dans le foie qui joue un rôle de transporteur dans le sang. Dans l’amylose TTR, ce tétramère se dissocie en monomères, qui eux-mêmes peuvent se réassembler en oligomères (après avoir subi une dénaturation partielle), puis en filaments et enfin en fibrilles amyloïdes. Le tafamidis joue un rôle de stabilisateur de ce tétramère en se liant aux deux sites de fixation de la thyroxine, ce qui empêche le processus dissociation en monomères et in fi ne ralentit la formation de dépôts amyloïdes.
La principale étude qui a justifié l’obtention de l’AMM européenne du tafamidis pour les cardiomyopathies amyloïdes est ATTR-ACT. Cet essai, randomisé versus placebo, international de phase 3, publié dans le NEJM en août 2018, a montré un effet clinique important sur des paramètres robustes. Étaient inclus des patients avec cardiomyopathie TTR associée à une insuffisance cardiaque sélectionnés sur les critères suivants : épaisseur pariétale ventriculaire gauche supérieure à 12 mm, insuffisance cardiaque congestive clinique, élévation du NT-proBNP > 600 pg/mL et biopsie montrant des dépôts amyloïdes. La population étudiée comprenait 441 patients (âge médian 74 ans, 90% d’hommes, #90% en classe NYHA II/III), dont 24% avec hATTR et 76% avec wtATTR.
Cet essai a montré une réduction relative de 30% de la mortalité toutes causes dans le groupe tafamidis (à la dose de 20 à 80 mg/j) par rapport au placebo après 30 mois de traitement (les courbes de mortalité divergeant dès le 12 ème mois), correspondant à une réduction absolue de mortalité totale de 13,4% (29,5% vs 42,9%). Les hospitalisations pour causes cardiovasculaires et pour insuffisance cardiaque étaient également réduites de 32 et 52%, respectivement. Dans le groupe tafamidis, il a été observé une baisse significativement moins importante de la capacité fonctionnelle évaluée par un test de marche de 6 minutes et d’un score de qualité de vie (Kansas City Cardiomyopathy Questionnaire–Overall Summary). Le profil de tolérance du tafamidis était bon avec un taux d’effets secondaires similaire à celui du groupe placebo, ce qui est concordant avec les données antérieures à l’étude (issues de l’utilisation de ce traitement dans la neuropathie amyloïde) ou postérieures (en RTU pour les cardiomyopathies TTR).
L’utilisation du tafamidis de manière concrète : pour qui, comment, à quel prix ?
Dans les suites de la publication de l’étude ATTR-ACT, le tafamidis (à la dose de 20mg/j) a d’abord obtenu une recommandation temporaire d’utilisation (RTU) dans l’indication « traitement de l’amylose cardiaque à transthyrétine de forme héréditaire ou sénile, chez les patients adultes présentant une insuffisance cardiaque restrictive de classe NYHA I, II ou III ». Dans ce cadre, il a été soumis à prescription hospitalière par des cardiologues « spécialisés dans la prise en charge des amyloses cardiaques ». Dans les suites de l’AMM européenne obtenue en février dernier, l’évaluation du traitement par la commission de transparence pour évaluer le service médical rendu et l’amélioration du service médical rendu n’a pas été effectuée à ce jour, de même que l’attribution du prix par le comité économique des produits de santé qui en découle. En guise de référence, le coût annuel du tafamidis à la dose de 20 mg utilisé pour le traitement de la neuropathie amyloïde est de l’ordre de 50 000 . Les modalités de prescription du tafamidis dans les suites de sa mise sur le marché ne sont pas non plus connues. La date à laquelle sera effectué le relais avec la RTU qui peut être raisonnablement attendue début 2021.
La question du coût est un point important dans un contexte où le développement de thérapies « innovantes » (immunothérapies, thérapie génique, oligonucléotides antisens, enzymothérapies, etc.) ciblant souvent des maladies rares ou avec des effectifs de patients restreints connait un développement exponentiel. Beaucoup de ces traitements ont un potentiel majeur pour améliorer la prise en charge de nos patients et leur émergence traduit des percées scientifiques importantes de la recherche académique et industrielle. Le développement de ces thérapies traduit également un repli stratégique de l’industrie pharmaceutique à la recherche de nouveaux modèles économiques avec des médicaments à population cible restreinte mais coût élevé faisant suite à la crise de celui des blockbusters. Notre société et nos décideurs vont désormais devoir réfléchir collectivement à la manière dont nos systèmes de santé pourront pouvoir supporter le coût croissant de ces nouveaux traitements. Le tafamidis figure sans doute parmi ceux pour lesquels le bénéfice clinique potentiel est le moins discutable.
Conclusion
Le tafamidis est un traitement spécifique de l’amylose cardiaque à transthyrétine ayant démontré un effet bénéfique fonctionnel et pronostique, chez les patients atteints de cette pathologie extrêmement sévère. Il est actuellement disponible en France dans cette indication dans le cas d’une RTU, et a récemment obtenu une AMM européenne et est en cours d’évaluation par la commission de transparence. Ces éléments viennent renforcer l’importance d’évoquer le diagnostic de cette pathologie devant une cardiomyopathie hypertrophique et/ou restrictive ou de manière plus large une insuffisance cardiaque à fraction d’éjection préservée, notamment (mais pas seulement) chez le sujet âgé.
N°35 – Sept 2020
Karim Wahbi1 ,
Service de cardiologie, hôpital Cochin, Université de Paris, Paris Cardiomyopathies Centre
Albert Hagège
Département de cardiologie, hôpital Européen Georges Pompidou, Université de Paris, Paris Cardiomyopathies Centre
L’auteur déclare ne pas avoir de liens d’intérêt
RÉFÉRENCES
1. Gonzalez-Lopez et al. 2015. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015 Oct 7;36(38):2585-94
2. Damy et al. 2016. Prevalence and clinical phenotype of hereditary transthyretin amyloid cardiomyopathy in patients with increased left ventricular wall thickness. Eur Heart J. 2016 Jun 14;37(23):1826-34
3. Castaño A, Narotsky DL, Hamid N, et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J. 2017;38:2879-2887.
4. Pereira NL, Grogan M, Dec GW. Spectrum of restrictive and infi ltrative cardiomyopathies: Part 2 of a 2-Part Series. J Am Coll Cardiol. 2018 Mar 13;71(10):1149-1166.
5. Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation. 2016 Jun 14;133(24):2404-12.
6. Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007-1016.