Le test génétique en cardiologie : chez qui, quand et pourquoi ?
Depuis une quinzaine d’années, le développement des techniques de génétique moléculaire a accéléré la disponibilité de ces tests dans de nombreux centres. Les tests génétiques dans le contexte des cardiomyopathies ont fait l’objet des 2010 de recommandations spécifiques par la Société Européenne de Cardiologie. (1) Ces tests ont maintenant toute leur place dans l’arsenal diagnostique et parfois pronostique du cardiologue dans de nombreuses situations cliniques. (2–4)
L’objet de cet article est de rappeler les principaux mécanismes génétiques qui sous-tendent les maladies cardiaques héréditaires, de détailler quelles sont les indications et les objectifs du test génétique en 2022.
Ce que le cardiologue doit savoir
La très grande majorité des maladies cardiaques héréditaires sont des maladies monogéniques autosomiques dominantes : une anomalie (= un variant ou une mutation) dans un seul des deux allèles d’un gène suffit à engendrer la maladie. Il existe deux exceptions à cela : la maladie de Fabry et les dystrophies de Duchenne ou Becker (comportant parfois une atteinte cardiaque) sont toutes trois des maladies récessives liées à l’X. C’est-à-dire qu’il existe une transmission de femme à homme avec des hommes atteints et des mères saines, avec parfois des femmes “porteuses” qui peuvent développent la maladie (bien que généralement à un âge plus avancé).
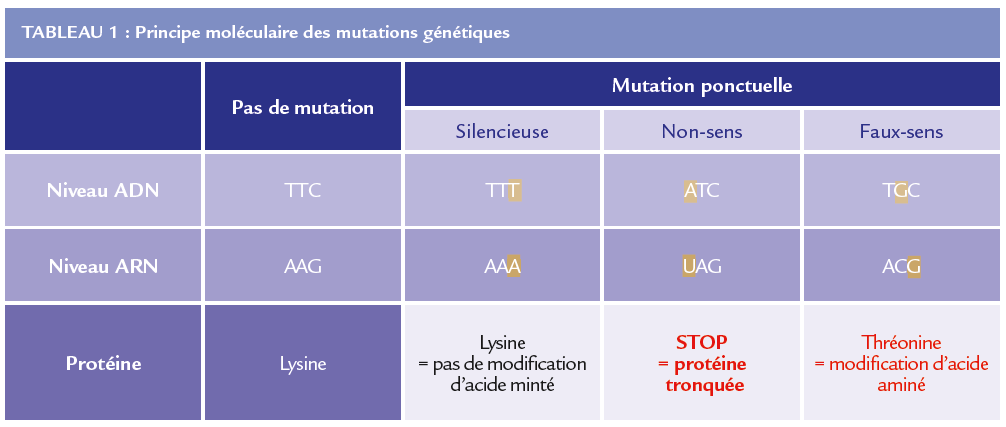
Les maladies cardiaques héréditaires sont décrites comme sporadiques : c’est-à-dire qu’elles n’atteignent pas toute la famille. Certains parents ou frères et sœurs sont indemnes de la maladie : soit en raison de sa pénétrance incomplète soit en raison d’une néomutation (le cas index est le premier cas de la famille présentant la mutation ce qui est bien plus rare). La nature de la mutation est variable : mutations faux-sens (modification d’acidité aminé modifiant les propriétés de la protéine), décalage du cadre de lecture, non-sens (modification d’acide aminé induisant un codon stop : protéine tronquée), petites délétions ou insertions et plus rarement grandes délétions. La mutation peut survenir sur toute la longueur d’un gène, sans localisation prédominante.
L’un des principaux défi s à relever pour établir un diagnostic génétique chez les patients atteints de maladies cardiaques héréditaires est l’hétérogénéité génétique considérable de la plupart des pathologies : toutes associées à de nombreuses mutations dans plusieurs gènes. Par exemple, la cardiopathie hypertrophique est associée à plus de 400 mutations dans plus de 15 gènes. Ainsi, le test génétique du probant porte sur un panel de gènes : environ 70 dans la cardiomyopathie dilatée (CMD), 5 dans la cardiomyopathie hypertrophique (CMH), 5 dans le syndrome du QT long et du QT court, dans la dysplasie arythmogène du ventricule droit (DAVD), 2 dans les tachycardies ventriculaires catécholergiques et un seul dans le syndrome de Brugada : le gène SCN5A le plus fréquemment muté).
Si une mutation est trouvée dans un gène d’intérêt pour la maladie cardiaque testée, le résultat sera classé en 5 types selon les recommandations internationales pour le rendu de résultat génétique. (5) Les variants de classe 4 et 5 correspondent à des mutations pathogènes avec certitude ou avec une très haute probabilité pouvant être utilisés pour du diagnostic présymptomatique dans la famille. Les variants de classe 1 et 2 sont des mutations bénignes n’expliquant par la maladie cardiaque. Les variants de classe 3 sont dits « de signification indéterminée » : ils doivent faire réaliser des analyses de ségrégation dans la famille. L’analyse de ségrégation consiste à analyser d’autres cas si possibles atteints et non atteints dans la famille : on cherche si les patients malades sont bien porteurs du variants et les patients sains non porteurs.
Enfin, il est à noter que dans le domaine des maladies cardiaques héréditaires, la pénétrance est très variable : c’est à dire que la proportion de patients qui vont développer des anomalies cardiaques en lien avec leur mutation est difficilement prédictible. Au sein d’une même famille, certains patients présentant la mutation vont présenter une atteinte phénotypique très sévère (par exemple cardiomyopathie dilatée avec dysfonction biventriculaire conduisant à une greffe cardiaque à un âge jeune), et d’autres peuvent rester asymptomatique toute leur vie ou développer une forme mineure à un âge tardif.
Objectifs
Les objectifs du test génétique sont principalement de deux types :
- Pour le cas index : assoir le diagnostic phénotypique (par exemple dans le syndrome du QT long) et apporter des éléments pronostiques dans certaines cardiomyopathies. Le test génétique n’est en revanche pas un test visant à étiqueter des cardiopathies borderline (comme des CMH avec dilatation ou des CMD avec hypertrophie) : le phénotype doit être clairement établi avant l’analyse génétique. Concernant le rôle pronostique, les mutations de la lamine (gène LMNA) ou du phospholamban (PLN) sont par exemple de mauvais pronostic dans les CMD. Ces résultats peuvent conduire à une modification du rythme du suivi ou des sanctions thérapeutiques (intégration prochaine des mutations génétiques dans les scores de risque rythmiques de la CMH, augmentation du seuil de FEVG conduisant à l’implantation d’un défibrillateur en prévention primaire dans les CMD avec certaines mutations délétères).
- Pour les apparentés : en cas de mutation retrouvée chez le cas index avec un niveau de certitude suffisant (variant de classe 4 ou 5), l’on peut réaliser du diagnostic présymptomatique chez les apparentés du 1er degré. Ainsi, les enfants (au-delà de 16 ans ou 3 ans avant le début de la maladie cardiaque chez le probant) et les membres de la fratrie du patient doivent bénéficier d’un test génétique. Les parents du probant peuvent également bénéficier du test génétique pour guider la poursuite des explorations : la branche familiale du parent porteur doit ensuite être testée (par exemple les oncles et tantes paternelles si la mutation est transmise par le papa). Si un membre de la famille n’est pas porteur, par exemple le frère du patient, les enfants de cet individu ne devront pas être testés puisque la mutation n’a pas pu leur être transmise (pas de saut de génération). En revanche, si la sœur du patient est atteinte par exemple, ses enfants (neveux et nièces du probant) devront également être testés.
Le résultat du test conditionne ensuite le rythme du suivi cardiologique. (Figure 1)
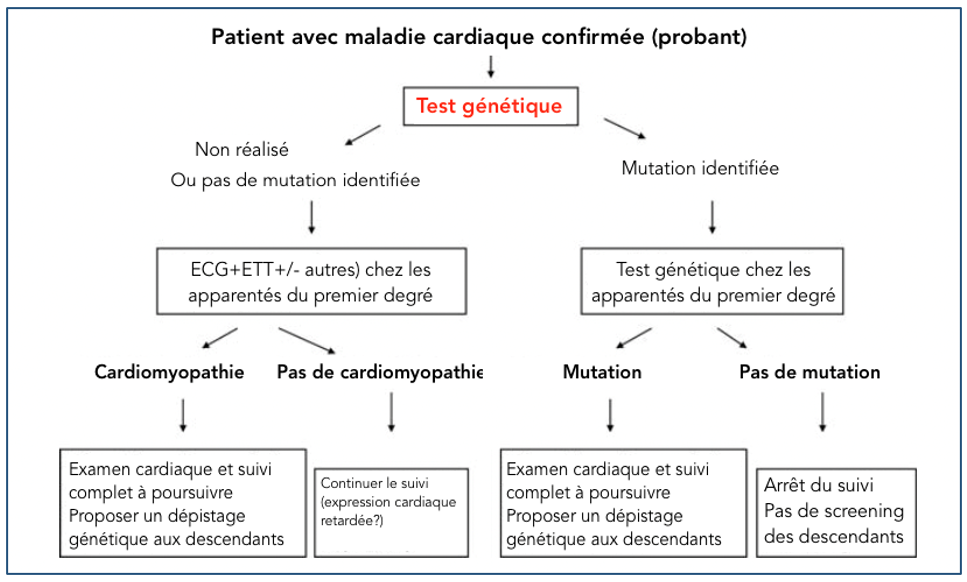
Figure 1 : stratégie diagnostique et suivi
En l’absence du variant familial chez un apparenté asymptomatique : le suivi peut être arrêté. En présence du variant familial chez un apparenté asymptomatique : l’apparenté doit bénéficier d’un bilan cardiaque de première intention puis d’un suivi régulier même en l’absence d’anomalie initiale (l’expression peut être retardée).
Dans certaines maladies cardiaques très sévères (CMD syndromiques avec myopathie par exemple), un diagnostic pré-natal ou pré-implantatoire peut être proposé. Si le bilan génétique ne retrouve pas de mutation chez le probant, cela ne remet pas en cause le diagnostic qui reste clinique. Ainsi, les apparentés doivent poursuivre un suivi cardiologique régulier même en l’absence de mutation chez le cas index.
Si le bilan génétique ne retrouve pas de mutation chez le probant, cela ne remet pas en cause le diagnostic qui reste clinique. Ainsi, les apparentés doivent poursuivre un suivi cardiologique régulier même en l’absence de mutation chez le cas index.
Indications
- Dans les cardiomyopathies :
En raison de la forte association des CMH et DAVD avec une anomalie génétique, le test génétique doit être systématique chez ces patients. Dans les CMD et les cardiomyopathies restrictives, le recours au test génétique doit être proposé en cas d’associations phénotypiques notables (par exemple CMD + myopathie ou CMD + troubles conductifs) ou s’il existe au moins un autre cas de cardiomyopathie ou de mort
subite suspecte dans la famille.
- Dans les morts subites récupérées :
Chez les patients ayant présenté un arrêt cardiaque extrahospitalier de cause cardiaque suspectée, un premier bilan doit être réalisé (ECG, ETT, IRM cardiaque et/ou autopsie si décès). Si ce bilan est normal et/ou s’il existe d’autres cas de mort subite suspectes dans la famille, le test génétique est recommandé. Le test génétique doit être adapté aux données phénotypiques retrouvées (panel de CMH, CMD ou selon le phénotype). En l’absence de données phénotypiques pouvant orienter le diagnostic dans une mort subite sur rythme choquable, il s’agit d’une « fibrillation ventriculaire idiopathique ». Dans ce cas, le laboratoire adaptera le panel de gènes analysés au bilan de ces morts subites rythmiques.
- Dans les maladies cardiaques rythmiques :
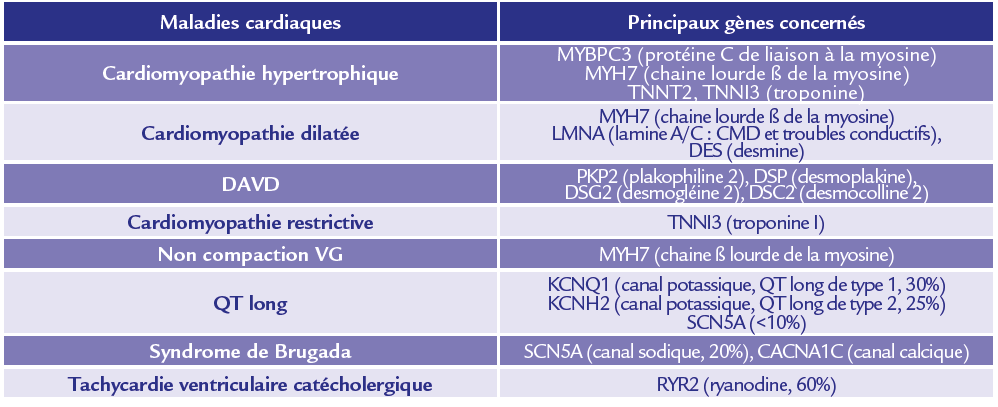
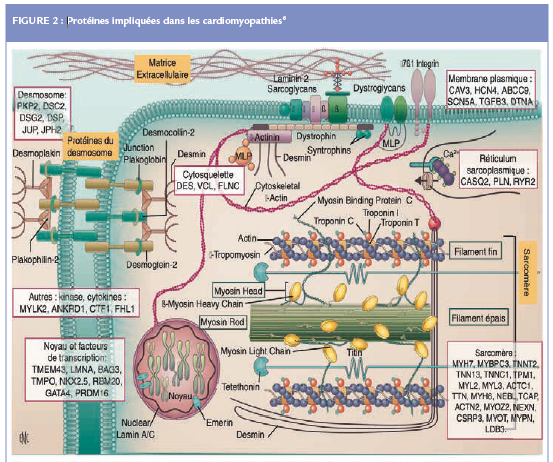
Le bilan génétique doit être systématique en cas de suspicion d’un syndrome du QT long congénital et de syndrome de Brugada (spontané ou provoqué). Dans les maladies rythmiques plus rares (QT court, tachycardie ventriculaire catécholergique), le bilan génétique doit également être systématique. Il existe un overlap génétique important dans la DAVD, la CMD et les formes frontières dites de cardiomyopathies arythmogènes (anomalies VG et hyperexcitabilité ventriculaire sans critère ventriculaire droit pour une DAVD). Les gènes partagés sont notamment LMNA (lamine A/C), SCN5A (canal sodique), DSP (desmoplakine), PLN (phospholamban) et DES (desmine). Les principaux gènes responsables de maladies cardiaques héréditaires sont présentés ci-dessous. Les sites d’actions des protéines codées par ces gènes sont schématisés dans la Figure 2.
Réalisation pratique
Le test génétique ne peut pas être réalisé sans le consentement éclairé écrit du patient après une information claire, loyale, appropriée par un praticien en capacité de recevoir, d’interpréter et d’expliquer les résultats du test génétique. Le consentement écrit doit être conservé dans le dossier, avec un double au laboratoire de génétique moléculaire et un double remis au patient. En cas de patient décédé (mort subite), les ayants-droits peuvent donner leur accord pour une analyse post-mortem, en accord avec les convictions du défunt.
Le prélèvement consiste en une simple prise de sang dans un tube EDTA, adressé à un laboratoire pratiquant des analyses de Cardiogénétique. Il est important de joindre au prélèvement un courrier expliquant les caractéristiques phénotypiques du patient, un électrocardiogramme et un arbre généalogique. Le résultat du test génétique doit être rendu par le praticien ayant prescrit le test, si possible de visu ou en téléconsultation (pas de rendu par mail, courrier ou téléphone !). En cas d’anomalie, il convient systématiquement de confirmer le résultat sur un 2e prélèvement.
Quelles informations donner à mon patient ?
- La probabilité que la maladie cardiaque soit d’origine génétique : quasi certain dans la CMH sans cause secondaire, le syndrome du QT long (après élimination des causes iatrogènes et circonstancielles), le syndrome de Brugada et la DAVD. 30% environ des CMD sont considérées comme d’origine génétique, ce qui augmente significativement dans les CMD associées à des troubles conductifs ou à une myopathie. Les causes génétiques sont très fréquentes dans la cardiomyopathie restrictive et la non-compaction ventriculaire gauche. En parallèle, la rentabilité du test génétique (probabilité de retrouver effectivement un variant causal en l’état actuel de la science) chez le probant est la suivante : 40-70% dans les CMH, ≤20% dans les CMD (sauf > 50% si CMD avec autre phénotype associé), 30-60% dans les DAVD, <20% dans les non-compaction ventriculaire gauche et les cardiomyopathies restrictives. Cela reflète bien le caractère encore partiel de nos connaissances des gènes responsables de maladies cardiaques héréditaires !
- Le mode de transmission et donc le risque de transmettre l’anomalie au sein de la famille (y compris la descendance) : dans la plupart des cas, l’anomalie est autosomique dominante et chaque enfant d’un individu présentant le variant a un risque de 50% d’avoir hérité de ce dernier.
- Les manifestations cliniques de la maladie et son histoire naturelle (y compris une éventuelle expression cardiaque tardive) : dépendant de chaque maladie cardiaque. A noter qu’un premier examen cardiaque normal chez un apparenté n’exclut pas la possibilité
que celui-ci soit porteur « sain » de la mutation et puisse exprimer la maladie plus tard : le suivi cardiologique est donc impératif ! - Le délai pour obtention des résultats : entre 8 et 12 mois pour le cas index (étude d’un panel de gènes) et 4-8 semaines pour les apparentés (on sait déjà quelle mutation rechercher ce qui accélère les analyses).
- Les intérêts du dépistage cardiaque au sein de la famille : si possible remettre une information écrite spécifique au patient pour qu’il transmette l’information à ces proches. Légalement, le patient est tenu d’informer ses apparentés de l’existence d’une mutation génétique dans la famille et de la nécessité de consulter pour un dépistage cardiaque et/ou génétique.
- Les coordonnées d’associations de patients et les sources d’information médicales (par exemple Orphanet ou fiches d’information de la filière Cardiogen). Si besoin, donner l’adresse d’un centre de référence pour le diagnostic moléculaire de la pathologie cardiaque.
En cas de mutation retrouvée chez un apparenté asymptomatique, il n’est pas obligatoire de prévenir les compagnies d’assurance en cas de prêt, de complémentaire santé, etc… tant qu’il n’existe aucune atteinte phénotypique exprimée. La loi protège la confidentialité des données génétiques et interdit aux employeurs et assureurs de demander la communication de ces résultats.
Conclusion
Le test génétique fait maintenant partie intégrante de la prise en charge globale du patient atteint d’une cardiomyopathie ou d’une canalopathie. Les indications du test génétique sont larges si et seulement si le phénotype est bien établi. Le prélèvement doit être réalisé systématiquement dans la CMH, la DAVD, le syndrome du QT long, de Brugada ou après une mort subite avec un bilan étiologique de première intention négatif. Il doit également être envisagé dans les CMD présentant certaines associations phénotypiques (myopathies, troubles conductifs) ou dans les familles présentant plusieurs membres atteints de CMD. La liste des centres de compétences et de références de cardio-génétique est disponible sur le site de la filière Cardiogen (http://www.cardiogen.aphp.fr).
Références
1. Charron P, Arad M, Arbustini E, Basso C, Bilinska Z, Elliott P, et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. European Heart Journal 2010;31:2715–26. https://doi.org/10.1093/eurheartj/ehq271.
2. Writing Committee Members, Ommen SR, Mital S, Burke MA, Day SM, Deswal A, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2020;142. https://doi.org/10.1161/CIR.0000000000000937.
3. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014;35:2733–79. https://doi.org/10.1093/eurheartj/ehu284.
4. Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2018;72:e91–220. https://doi.org/10.1016/j.jacc.2017.10.054.
5. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 2015;17:405–24. https://doi.org/10.1038/gim.2015.30.
6. P. Richard, F. Ader, P. Charron, Génétique des cardiomyopathies héréditaires, Volume, Issue, /2018, Pages, ISSN 1166-4568, http://dx.doi.org/10.1016/S1166-4568(18)53103-X (http://www.sciencedirect.com/science/article/pii/S1166-4568(18)53103-X)
Orianne Weizman, Paris