
En août 2023, sont parues les nouvelles recommandations européennes sur les cardiomyopathies1 . Elles résultent d’un effort de la Société Européenne de Cardiologie pour synthétiser les connaissances sur le domaine. Jusqu’à présent, nous avons connu des recommandations spécifiques sur la cardiomyopathie hypertrophique2, l’amylose3 et la dysplasie arythmogène du ventricule droit, il s’agit ici de la première tentative d’unification des recommandations. Si ces nouvelles recommandations sont novatrices à plusieurs égards, un des éléments importants est la création d’un nouveau groupe de cardiomyopathies : la cardiomyopathie non dilatée du ventricule gauche (CNDVG). Il s’agit d’une nouvelle famille que cet article se propose de détailler.
Généralités
Pour la première fois, ces recommandations mettent l’accent sur une approche globale, multidisciplinaire et multimodale des cardiomyopathies. Elles détaillent des parcours de soins précis pour les patients, de l’enfance à l’âge adulte, y compris des situations particulières comme la grossesse. Elles mettent également l’accent sur une approche diagnostique phénotypique où l’IRM cardiaque devient un examen incontournable. Enfin, elles évoquent la place grandissante de la génétique.
Définition de la cardiomyopathie
Une cardiomyopathie peut être définie par un désordre myocardique entrainant une anomalie structurelle et fonctionnelle du cœur et ce, en l’absence de coronaropathie, d’HTA, de pathologie valvulaire ou d’anomalie congénitale significative. Les anomalies morphologiques sont définies par la présence d’une hypertrophie ou d’une dilatation d’un ou des deux ventricules ou d’anomalies de signal non ischémiques sur des séquences IRM de caractérisation tissulaire. Quant aux anomalies fonctionnelles, elles sont de deux types : une dysfonction systolique segmentaire ou globale d’un ou des deux ventricules et/ou une dysfonction diastolique d’un ou des deux ventricules. On voit ici l’apport essentiel de l’échocardiographie comme examen de première intention et celui de l’IRM cardiaque comme examen de deuxième intention.
Approche phénotypique des cardiomyopathies
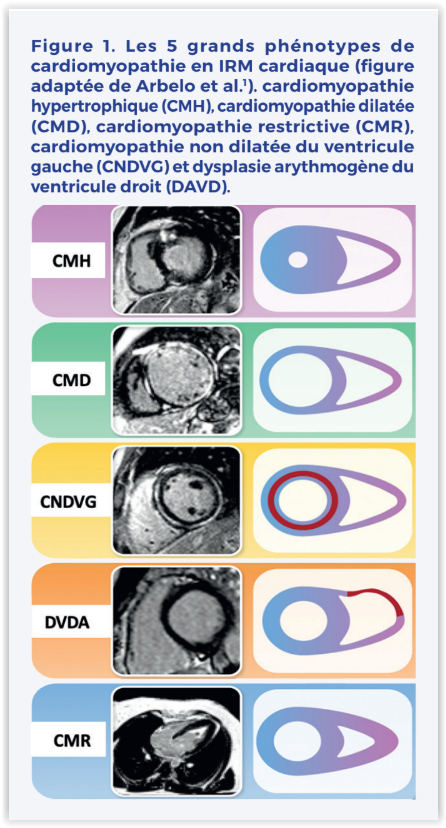
Une des nouveautés de ces recommandations est l’approche phénotypique. Grossièrement, le cardiologue suspectera une cardiomyopathie sur des scenarii cliniques variables (symptômes, anomalies ECG, dépistage familial…). Il confirmera ensuite l’existence d’anomalies myocardiques morphologiques et fonctionnelles en échocardiographie et orientera le patient vers la réalisation d’une IRM cardiaque qui permettra idéalement de classer la cardiomyopathie dans un des 5 phénotypes suivants : cardiomyopathie hypertrophique (CMH), dilatée (CMD), restrictive (CMR), non dilatée du ventricule gauche (CNDVG) et dysplasie arythmogène du ventricule droit (DAVD). (Figure 1)
Cardiomyopathie non dilatée du ventricule gauche (CNDVG) : une nouvelle famille
Définition : Selon les recommandations, le phénotype de CNDVG est défini soit par :
– la présence de zones de cicatrices non ischémiques du VG ou de remplacement graisseux de myocytes, indépendamment de la présence de troubles de la cinétique segmentaire ou globale.
– soit par une hypokinésie globale isolée du VG sans cicatrice.
1. Arbelo E et al. 2023 ESC Guidelines for the management of cardiomyopathies. European Heart Journal 2023 Oct 1;44(37):3503-3626.
2. Elliott P et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. European Heart Journal 2014 35, 2733–2779.
3.Garcia-Pavia P et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases.
European Heart Journal 2021 42, 1554–1568.
Cette définition appelle plusieurs remarques. Premièrement, il s’agit d’une définition assez floue qui nécessitera des précisions dans le futur. En effet, les cardiologues pratiquant l’échographie cardiaque connaissent la difficulté de juger de la cinétique segmentaire ou globale du VG, cette analyse étant dépendante de l’expérience du praticien, des conditions de charge et des situations cliniques.
Deuxièmement, cette définition inclut une notion de caractérisation tissulaire myocardique ce qui rend l’IRM cardiaque incontournable et implique que le clinicien soit familiarisé avec les séquences d’IRM cardiaque. Troisièmement, parler de « zones de cicatrices non ischémiques du VG ou de remplacement graisseux de myocytes » va probablement impliquer une place grandissante de la biopsie myocardique dans l’exploration de ce phénotype. Enfin, cette définition n’inclut aucune précision ni sur le niveau de fraction d’éjection ventriculaire gauche ni sur les volumes du VG dont on sait que les valeurs normales diffèrent selon la technique utilisée (IRM ou échocardiographie).
Rappels sur les outils IRM de caractérisation tissulaire :
Les outils disponibles en IRM pour caractériser le myocarde sont actuellement les cartographies T1, T2, T2* , le volume extracellulaire (ECV) et le réhaussement tardif (RT). Nous détaillons ici uniquement le T1, l’ECV et le RT.
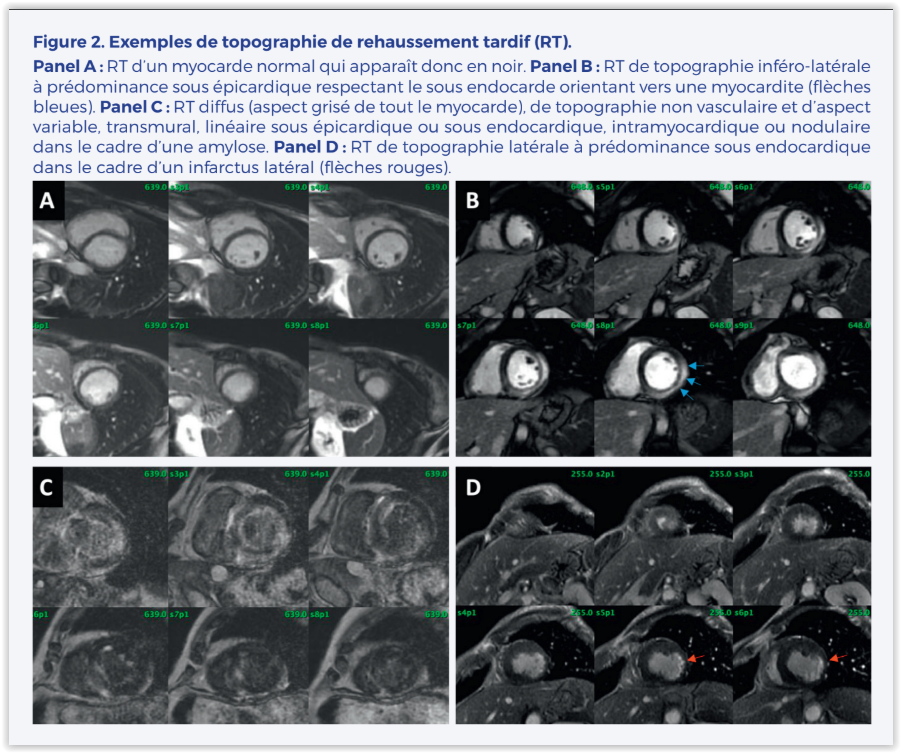
– Séquences de réhaussement tardif (RT) : Parmi les outils dont on dispose en IRM cardiaque pour caractériser le tissu myocardique, le RT est très certainement le plus célèbre. Il s’agit d’une imagerie réalisée à distance de l’injection de gadolinium (classiquement 10 minutes après l’injection). Le gadolinium est un traceur à distribution extracellulaire qui a totalement quitté le myocarde normal à ce timing, le muscle normal apparaît donc totalement noir. Si du gadolinium est encore présent à 10 minutes de l’injection, il s’agit d’une situation pathologique. Le RT est souvent considéré à tort comme de la fibrose myocardique. En réalité, il peut s’agir de dépôts amyloïdes, d’eau (œdème/inflammation) ou de fibrose. En effet, lorsqu’il existe une maladie myocardique quelle qu’elle soit, celle-ci va être responsable de la mort et donc de la disparition de myocytes, le volume extracellulaire va donc augmenter. Le gadolinium injecté va venir s’y déposer et y rester piégé. Le RT va se traduire par un hypersignal (blanc) en comparaison du myocarde sain qui apparaît en hyposignal (noir). En cas de maladie diffuse du myocarde, le dépôt se faisant de manière homogène, les séquences de RT seront moins parlantes voire normales par manque de contraste entre myocarde sain et myocarde pathologique. L’absence de RT n’est donc pas forcément synonyme d’absence de fibrose. En revanche, une augmentation localisée du secteur extracellulaire se traduira par un hypersignal et c’est la topographie de cet hypersignal qui va nous orienter vers une cause plutôt qu’une autre (ischémique/ myocardite/amylose…) (Figure 2).
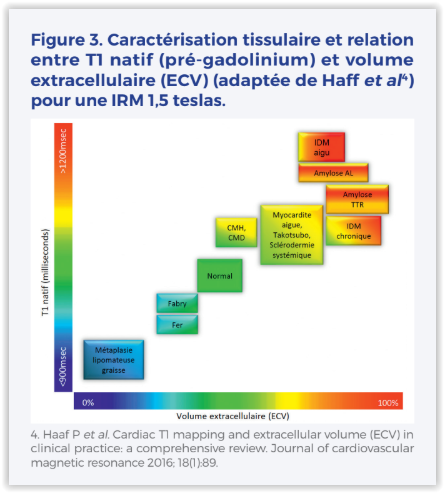
– Cartographie T1 et mesure du volume extracellulaire (ECV) (Figure 3) Le T1 mapping et l’ECV sont des techniques avancées d’IRM cardiaque utilisées pour caractériser les tissus myocardiques de manière plus précise et non invasive. Le T1 mapping permet de mesurer le temps de relaxation longitudinale (T1) du myocarde, fournissant ainsi des informations sur ses propriétés biochimiques et biophysiques. Même si cette valeur augmente avec l’âge et varie sensiblement selon l’intensité du champ magnétique et le constructeur, le lecteur peut retenir très schématiquement une valeur normale entre 850ms et 1050/1100ms pour un champ magnétique de 1,5 Teslas.

Cette mesure réalisée avant injection de gadolinium, génère des cartes de relaxation T1 classiquement sur des coupes petit axe du myocarde. On obtient ainsi une mesure qui nous oriente vers la composition du myocarde (muscle normal ou au contraire teneur en fer, graisse, eau, dépôts amyloïdes ou fibrose). Un T1 bas fait ainsi évoquer deux maladies : la maladie de Fabry et l’hémochromatose. À l’inverse, un T1 élevé orientera vers d’autres pathologies (CMH, CMD, myocardite, amylose…) (Figure 3). Cependant, le T1 natif ne fait pas la différence entre le muscle myocardique d’une part et les vaisseaux et le sang circulant à l’intérieur d’autre part. C’est tout l’intérêt de l’ECV qui permet de s’affranchir du signal T1 du sang. L’ECV est quantifié à partir des valeurs de T1 avant et après l’administration de gadolinium. Il représente la proportion du volume myocardique occupé par l’espace extracellulaire, reflétant ainsi la présence de fibrose diffuse ou d’autres modifications interstitielles telles que l’œdème ou les dépôts amyloïdes par exemple. L’ECV est particulièrement utile pour évaluer l’étendue et la distribution de la fibrose myocardique. Schématiquement, on pourra retenir une valeur anormale d’ECV >30%.
Quelles sont les caractéristiques de cette nouvelle famille ?
Puisqu’il s’agit d’une nouvelle famille, nous ne disposons pas de données épidémiologiques spécifiques. La prévalence est, pour le moment, inconnue, d’autant qu’il existe forcément un recouvrement avec d’autres phénotypes dans leurs formes débutantes ou frustes (ex : l’amylose débutante). De plus, la coexistence d’autres maladies fréquentes (HTA, coronaropathie non sévère, rétrécissement aortique modéré ou moyen…) sont autant de facteurs qui vont rendre la tâche du clinicien difficile.
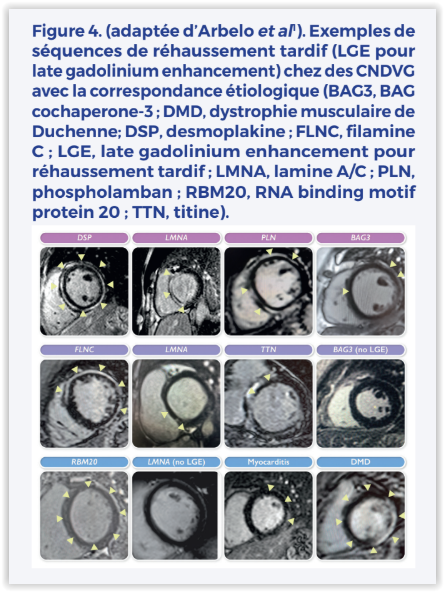
Cette nouvelle famille étant souvent associée à des mutations, le clinicien devra avoir le réflexe d’adresser le patient pour une analyse génétique. Il est également possible que, dans un futur proche, soient identifiés des patterns IRM orientant vers une mutation plutôt qu’une autre (Figure 4).
Depuis ces recommandations, quelques études ont été publiées sur cette nouvelle famille. Ainsi Eda et al5 ont pu montrer, sur une cohorte rétrospective entre 2004 et 2018, une absence de différence significative sur un critère composite d’évènements cardio-vasculaires associant décès par insuffisance cardiaque, mort subite et implantation d’une assistance entre les CNDVG et les CMD.
De même, Castrichini et al6 , ont pu montrer à partir d’une cohorte multicentrique (4 centres en Europe et aux USA) de 462 patients dont 227 patients avec CMD et 235 avec CNDVG que la topographie septale du RT et la dilatation du VG étaient associées à un risque accru de mort subite et d’arythmie ventriculaire grave, les CNDVG sans RT septal apparaissant comme le groupe le moins à risque et les CMD avec RT septal comme le groupe le plus à risque, tandis que les CMD sans RT septal et les CNDVG avec RT septal constituent des groupes à risque intermédiaire. Ces résultats nécessitent confirmation et affinement sur des cohortes plus conséquentes et mieux définies.
Une place grandissante de la génétique :
Lorsque le phénotype de CNDVG est identifié, et de manière générale pour les cardiomyopathies, une enquête familiale est indispensable. Elle doit être scrupuleuse, complète et souvent répétée. En effet, il n’est pas rare que les patients se souviennent à posteriori d’histoires familiales après y avoir réfléchi ou avoir questionné leurs proches.
L’analyse génétique permettra de caractériser la cardiomyopathie, d’organiser un conseil génétique et un dépistage des apparentés, d’aider à la stratification pronostique (certaines anomalies génétiques comme les laminopathies étant associées à un pronostic plus défavorable), d’aider à la décision d’implantation de défibrillateur dans le cas des mutations associées avec un très haut risque rythmique.
Dans le futur, il est très probable que le champ de la génétique s’étende encore. Par exemple, Garcia-Pavia et al7 ont pu montrer qu’une mutation asymptomatique de la titine prédisposait les patients atteints de cancer et devant recevoir une chimiothérapie potentiellement cardiotoxique à développer une cardiotoxicité.
Cas clinique illustratif :
Il s’agit d’une patiente de 57 ans sans antécédent notable ni facteur de risque cardio-vasculaire, aucun antécédent familial au terme du premier interrogatoire réalisé aux soins intensifs. Elle a présenté un épisode de douleur thoracique 1 an auparavant qui a fait réaliser un bilan cardiologique comprenant ECG, échocardiographie et épreuve d’effort, tous ces examens étaient normaux. Depuis, elle rapporte des palpitations fréquentes et des douleurs thoraciques atypiques intermittentes.
Alors qu’elle pratique un footing, elle va présenter une sensation de malaise, asthénie majeure, palpitations et dyspnée. Il s’ensuit une perte de connaissance brève. L’ECG réalisé par le SAMU retrouve une tachycardie régulière à 215bpm, QRS fins, mais avec une dissociation atrio-ventriculaire permettant de retenir le diagnostic de tachycardie ventriculaire (Figure 5).
5. Eda et al. Non-dilated left ventricular cardiomyopathy vs. dilated cardiomyopathy: clinical background and outcomes. ESC Heart Failure 2024; 11: 1463–1471.
6. Castrichini et al. Magnetic Resonance Imaging Characterization and Clinical Outcomes of Dilated and Arrhythmogenic Left Ventricular Cardiomyopathies. J Am Coll Cardiol 2024 May 14;83(19):1841-1851.
7. Garcia-Pavia P et al. Genetic Variants Associated With Cancer Therapy-Induced Cardiomyopathy. Circulation 2019 Jul 2;140(1):31-41.

Après échec du traitement médicamenteux, la patiente a eu un choc électrique après courte sédation. La Figure6 montre L’ECG après cardioversion. L’échographie cardiaque après retour en rythme sinusal montre un VG non dilaté, globalement hypokinétique avec une FEVG calculée à 50% sans autres anomalies. Un bilan étiologique biologique exhaustif ne montre aucune anomalie.
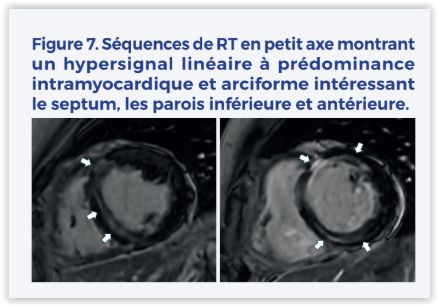
Elle a pu ensuite bénéficier d’une exploration électrophysiologique qui retrouvait des extrasystoles ventriculaires para-hissiennes avec deux foyers de sortie, un hissien droit et un septal haut gauche. Ces foyers ont été ablatés. Le bilan étiologique a comporté une échographie cardiaque réalisée à distance qui retrouve une fraction d’éjection normale contrastant avec une IRM cardiaque plutôt en faveur d’une fraction d’éjection aux alentours de 45% avec un VG non dilaté ainsi que de la fibrose arciforme centromyocardique du septum basal et moyen et de la paroi antérieure (Figure 7).
Le coroscanner retrouve une infiltration mixte de l’IVA proximale semblant significative non confirmée par la coronarographie. La reprise de l’interrogatoire a permis de retrouver deux décès suspects et brutaux avant 50ans ainsi que 2 apparentés porteurs de défibrillateur dans la famille.
Finalement, le dossier de la patiente a été discuté en staff rythmologique, la pose d’un défibrillateur a été décidé. Dans ce cas clinique, nous avons utilisé l’approche phénotypique prônée par les nouvelles recommandations. La patiente s’est présentée avec un scénario clinique associant des symptômes et des anomalies ECG. L’IRM a permis de classer la patiente dans le groupe des CNDVG. L’analyse génétique a permis de révéler que la patiente est porteuse d’un variant dans le gène LMNA (laminopathie). Ce cas clinique souligne également l’importance d’un interrogatoire répété qui aura laissé le temps à la patiente de questionner son entourage puisqu’il s’agissait ici d’une famille nombreuse dont les membres étaient éclatés dans le monde entier.
Conclusion
La reconnaissance de la CNDVG comme une nouvelle entité nosologique par la Société Européenne de Cardiologie en 2023 représente une avancée significative. Cette nouvelle classification permet une meilleure compréhension des formes de cardiomyopathie qui ne se manifestent pas par une dilatation ventriculaire gauche classique mais qui présentent néanmoins des anomalies myocardiques structurelles et fonctionnelles. L’échocardiographie et l’IRM cardiaque sont les deux outils essentiels à disposition du clinicien évaluant une cardiomyopathie. Le recours à l’analyse génétique va devenir incontournable lorsqu’une CNDVG est identifiée. À l’avenir, des recherches supplémentaires et des collaborations internationales seront essentielles pour affiner les critères diagnostiques, accumuler des données épidémiologiques, mieux comprendre le pronostic et préciser le risque rythmique et le risque évolutif vers l’insuffisance cardiaque.